Creationism in Crisis
Evolution or Malevolent Design? More Zoonomia Headaches for Creationists
Evolution or Malevolent Design? More Zoonomia Headaches for Creationists
Image: istock

The Zoonomia Project
The Zoonomia Consortium was formed in 2015 by a group of scientists from several institutions, including the University of California, Davis, the University of California, Berkeley, the University of California, Santa Cruz, and the Broad Institute of MIT and Harvard. The goal of the consortium was to develop a comprehensive database of animal genomes that could be used to study and compare the genetic basis of traits across different species.Yet more evidence of mammalian common origins and the lack of intelligence in design, or, if we assume design, then evidence of malevolence, has emerged from the Zonomia Consortium this week.
In 2020, the Zoonomia Consortium published a paper in the journal Nature describing the results of their work. The paper, entitled "Zoonomia Consortium, A comparative genomics multitool for scientific discovery and conservation," provides an overview of the consortium's approach and the tools they developed.
The Zoonomia database includes genomic data from over 100 species of animals, including mammals, birds, reptiles, and fish. The database is freely available to researchers and includes a variety of tools for analyzing and comparing genomic data, such as genome browsers, alignment tools, and gene expression analysis tools.
The Zoonomia Consortium's work has significant implications for conservation biology, as it allows researchers to compare the genetic basis of traits across different species and identify genetic factors that may be important for species survival. For example, researchers could use the Zoonomia database to identify genes that are associated with disease resistance or tolerance to environmental stressors, which could inform conservation efforts.
Reference:Lewin, H.A., Robinson, G.E., Kress, W.J. et al. Zoonomia Consortium, A comparative genomics multitool for scientific discovery and conservation. Nature 587, 240–245 (2020). https://doi.org/10.1038/s41586-020-2876-6chatGPT3 "Tell me about the Zoonomia Consortium, with references." [Response to user question]
Retrieved from: https://chat.openai.com/
Researchers at the University of Southern California (USC) Keck School of Medicine have used their database of the genomes of 240 different mammal species, to work out why humans suffer from diseases despite being the end-point of millions of years of evolution that might have been expected to weed out predisposition to diseases.
It turns out that most of the genes that lead to diseases are in a group of highly conserved genes that are common to all mammals. These genes appear to be highly constrained in evolutionary terms, meaning that any mutations would have been seriously deleterious so making their carriers less successful in competition with carriers of the normal allele.
As the USC Keck School of Medicine news release explains:
“Why do humans have disease if they went through millions of years of evolution?” It’s a question Steven Gazal, PhD, assistant professor of population and public health sciences at the Keck School of Medicine of USC, hopes to answer.More detail is included in the structures abstract to the team's paper published in the special Zoonomia edition of Science:
Gazal is part of an international team of researchers who have become the first to precisely identify base pairs of the human genome that remained consistent over millions of years of mammalian evolution, and which play a crucial role in human disease. The findings were published in a special Zoonomia edition of Science.
Gazal and his team analyzed the genomes of 240 mammals, including humans, zooming in with unprecedented resolution to compare DNA. They were able to identify base pairs that were “constrained” – meaning they remained generally consistent – across mammal species over the course of evolution. Individuals born with mutations on these genes may not have been as successful within their species or were otherwise not likely to pass down the genetic variation. “We were able to identify where gene mutations are not tolerated in evolution, and we demonstrated that these mutations are significant when it comes to disease,” explains Gazal.
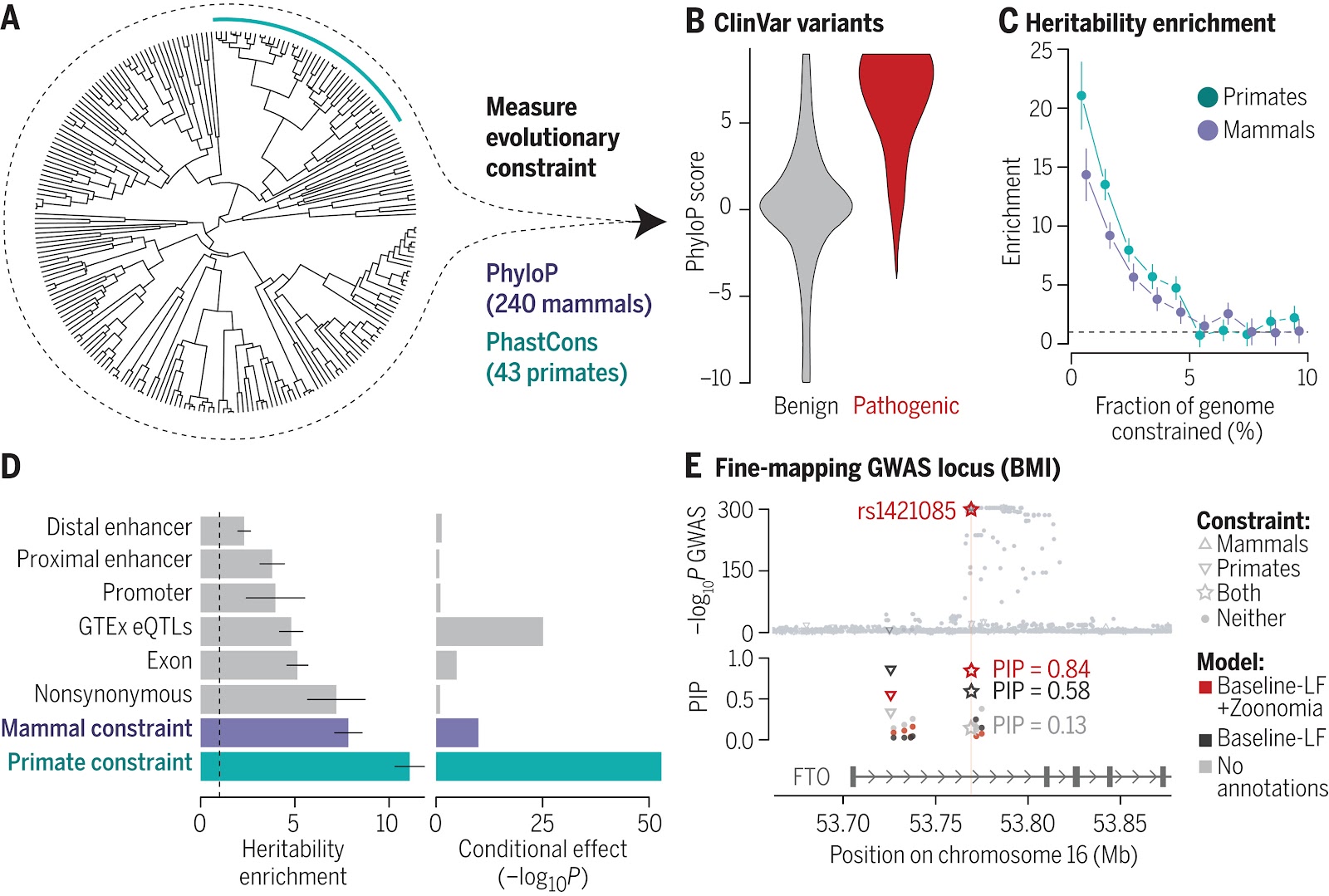
The team found that 3.3% of bases in the human genome are “significantly constrained,” including 57.6% of the coding bases that determine amino acid position, meaning these bases had unusually few variants across species in the dataset. The most constrained base pairs in mammals were over seven times more likely to be causal for human disease and complex trait, and over 11 times more likely when researchers looked at the most constrained base pairs in primates alone.
The dataset was provided by the Zoonomia consortium, which according to the project website, “is applying advances in DNA sequencing technologies to understand how genomes generate the tremendous wealth of animal diversity.” Gazal gives credit to Zoonomia for making this type of data available to researchers and anticipates it will be widely used by human geneticists. “It’s a cheap resource to generate, as opposed to datasets generated in human genetic studies,” says Gazal.
His team’s findings are a significant step forward, as Gazal notes, “we do not understand 99% of the human genome, so it is fundamental to understand which part has been constrained by evolution and is likely to have an impact on human phenotypes.” Their discoveries and methods could become crucial tools for further research.
The next step for Gazal and his team is to repeat the process with a primate-only dataset. By restricting the subjects, they hope to focus on functions of DNA that appeared more recently in human evolution. “We expect this to be even more useful in determining information on human disease,” says Gazal.
Structured AbstractThe significant points, that creationist will avoid like the plague, are:
INTRODUCTION
Thousands of genetic variants have been associated with human diseases and traits through genome-wide association studies (GWASs). Translating these discoveries into improved therapeutics requires discerning which variants among hundreds of candidates are causally related to disease risk. To date, only a handful of causal variants have been confirmed. Here, we leverage 100 million years of mammalian evolution to address this major challenge.
RATIONALE
We compared genomes from hundreds of mammals and identified bases with unusually few variants (evolutionarily constrained). Constraint is a measure of functional importance that is agnostic to cell type or developmental stage. It can be applied to investigate any heritable disease or trait and is complementary to resources using cell type– and time point–specific functional assays like Encyclopedia of DNA Elements (ENCODE) and Genotype-Tissue Expression (GTEx).
RESULTS
Using constraint calculated across placental mammals, 3.3% of bases in the human genome are significantly constrained, including 57.6% of coding bases. Most constrained bases (80.7%) are noncoding. Common variants (allele frequency ≥ 5%) and low-frequency variants (0.5% ≤ allele frequency < 5%) are depleted for constrained bases (1.85 versus 3.26% expected by chance, P < 2.2 × 10−308). Pathogenic ClinVar variants are more constrained than benign variants (P < 2.2 × 10−16). The most constrained common variants are more enriched for disease single-nucleotide polymorphism (SNP)–heritability in 63 independent GWASs. The enrichment of SNP-heritability in constrained regions is greater (7.8-fold) than previously reported in mammals and is even higher in primates (11.1-fold). It exceeds the enrichment of SNP-heritability in nonsynonymous coding variants (7.2-fold) and fine-mapped expression quantitative trait loci (eQTL)–SNPs (4.8-fold). The enrichment peaks near constrained bases, with a log-linear decrease of SNP-heritability enrichment as a function of the distance to a constrained base.
Zoonomia constraint scores improve functionally informed fine-mapping. Variants at sites constrained in mammals and primates have greater posterior inclusion probabilities and higher per-SNP contributions. In addition, using both constraint and functional annotations improves polygenic risk score accuracy across a range of traits. Finally, incorporating constraint information into the analysis of noncoding somatic variants in medulloblastomas identifies new candidate driver genes.
CONCLUSION
Genome-wide measures of evolutionary constraint can help discern which variants are functionally important. This information may accelerate the translation of genomic discoveries into the biological, clinical, and therapeutic knowledge that is required to understand and treat human disease.
Using evolutionary constraint in genomic studies of human diseases.
(A) Constraint was calculated across 240 mammal species, including 43 primates (teal line). (B) Pathogenic ClinVar variants (N = 73,885) are more constrained across mammals than benign variants (N = 231,642; P < 2.2 × 10−16). (C) More-constrained bases are more enriched for trait-associated variants (63 GWASs). (D) Enrichment of heritability is higher in constrained regions than in functional annotations (left), even in a joint model with 106 annotations (right). (E) Fine-mapping (PolyFun) using a model that includes constraint scores identifies an experimentally validated association at rs1421085. Error bars represent 95% confidence intervals. BMI, body mass index; LF, low frequency; PIP, posterior inclusion probability.
Abstract
Thousands of genomic regions have been associated with heritable human diseases, but attempts to elucidate biological mechanisms are impeded by an inability to discern which genomic positions are functionally important. Evolutionary constraint is a powerful predictor of function, agnostic to cell type or disease mechanism. Single-base phyloP scores from 240 mammals identified 3.3% of the human genome as significantly constrained and likely functional. We compared phyloP scores to genome annotation, association studies, copy-number variation, clinical genetics findings, and cancer data. Constrained positions are enriched for variants that explain common disease heritability more than other functional annotations. Our results improve variant annotation but also highlight that the regulatory landscape of the human genome still needs to be further explored and linked to disease.
Patrick F. Sullivan et al.,
Leveraging base-pair mammalian constraint to understand genetic variation and human disease.
Science 380, eabn2937 (2023). DOI:10.1126/science.abn2937
© 2023 The Authors. Published by American Association for the Advancement of Science.
Reprinted with kind permission under license #5553150959442
- The evidence of common ancestry of the mammals in the study.
- The lack of any evidence that the scientists were rejecting the TOE as an explanation for the observed facts.
- The inevitable conclusion that if a magic designer designed genes to cause diseases, then the designer can only be described as malevolent; it this was an accident, then incompetence is the only explanation.
- Even if, as 'theistic evolutionists' argue, an invisible supernatural deity was guiding human evolution, it was either incompetent, or malevolent, in allowing genes that caused diseases to be conserved in the evolving human genome.

No comments :
Post a Comment
Obscene, threatening or obnoxious messages, preaching, abuse and spam will be removed, as will anything by known Internet trolls and stalkers, by known sock-puppet accounts and anything not connected with the post,
A claim made without evidence can be dismissed without evidence. Remember: your opinion is not an established fact unless corroborated.